
Modelling Protein Interactions and Dynamics in Neuronal Signalling
Resources
Publications
- Yuan J-H, Bosco Han S, Richter S, Wade RC & Kokh DB (2019). Druggability Assessment in TRAPP using Machine Learning Approaches. J. Chem. Inf. Model.
- Bruce NJ, Narzi D, Trpevski D, van Keulen SC, Nair AG, Röthlisberger U, Wade RC, Carloni P & Hellgren Kotaleski J (2019). Regulation of adenylyl cyclase 5 in striatal neurons confers the ability to detect coincident neuromodulatory signals. PLOS Computational Biology.
- Bruce NJ, Ganotra GK, Richter S & Wade RC (2019). KBbox: A Toolbox of Computational Methods for Studying the Kinetics of Molecular Binding. Journal of Chemical Information and Modeling.
- Kokh DB, Kaufmann T, Kister B & Wade RC (2019). Machine Learning Analysis of τRAMD Trajectories to Decipher Molecular Determinants of Drug-Target Residence Times. Frontiers in Molecular Biosciences.
- Tong R, Wade RC & Bruce NJ (2019). Comparative electrostatic analysis of adenylyl cyclase for isoform dependent regulation properties. PROTEINS: Structure, Function, and Bioinformatics.
- Frezza E & Lavery R (2019). Internal Coordinate Normal Mode Analysis: A Strategy To Predict Protein Conformational Transitions. The Journal of Physical Chemistry B.
- Reille S, Garnier M, Robert X, Gouet P, Martin J & Launay G (2018). Identification and visualization of protein binding regions with the ArDock server. Nucleic Acids Research.
- Frezza E, Martin J & Lavery R (2018). A molecular dynamics study of adenylyl cyclase: The impact of ATP and G-protein binding. PLOS ONE.
- Bruce NJ, Ganotra GK, Kokh DB, Sadiq SK & Wade RC (2018). New approaches for computing ligand–receptor binding kinetics. Current Opinion in Structural Biology.
- van Keulen SC & Rothlisberger U (2017). Exploring the inhibition mechanism of adenylyl cyclase type 5 by N-terminal myristoylated Gαi1. PLOS Computational Biology.
- Casasnovas R, Limongelli V, Tiwary P, Carloni P & Parrinello M (2017). Unbinding Kinetics of a p38 MAP Kinase Type II Inhibitor from Metadynamics Simulations. Journal of the American Chemical Society.
- van Keulen SC & Rothlisberger U (2016). Effect of N-terminal myristoylation on the active conformation of Gαi1–GTP. Biochemistry.
- Tong R, Wade RC Bruce NJ (2016). Comparative electrostatic analysis of adenylyl cyclase for isoform dependent regulation properties. Proteins: Structure, Function, and Bioinformatics.
____________________________
 At the most fundamental level, senses, cognition and brain function ultimately depend on complex molecular processes occurring within neurons. From light-sensing proteins in the eye or proteins that respond to forces in the skin, through to the response of neurons in the brain, the way we are able to interact with our surrounding and learn from our experiences, is governed by the interactions of many 1000s of molecules.
At the most fundamental level, senses, cognition and brain function ultimately depend on complex molecular processes occurring within neurons. From light-sensing proteins in the eye or proteins that respond to forces in the skin, through to the response of neurons in the brain, the way we are able to interact with our surrounding and learn from our experiences, is governed by the interactions of many 1000s of molecules.
The increasing rate of production of biomolecular structural data in the last few decades means that we are currently in a position to investigate how many of these processes occur at the molecular level.
What makes molecular models special?
Physics-based molecular simulations offer a way to investigate these molecular processes, allowing us to gain mechanistic insights and predict parameters that can be used to constrain higher-level models of subcellular signalling. Additionally, molecular models can be used to help design drugs that can modulate these processes, allowing new treatments for brain-related diseases.
In the Brain Simulation Platform, we are developing computational tools and workflows to allow these simulations to be performed in a systematic way.
What are the specific questions we want to address in the HBP?
As a concrete example, one area we are focusing on is cellular signalling in dopamine 1 receptor (D1R)-containing medium spiny neurons (MSNs), which are a key mediator of reinforced learning in the striatum. As such, they are one of the cell types being simulated within the Brain Simulation Platform (see Signalling Cascades).
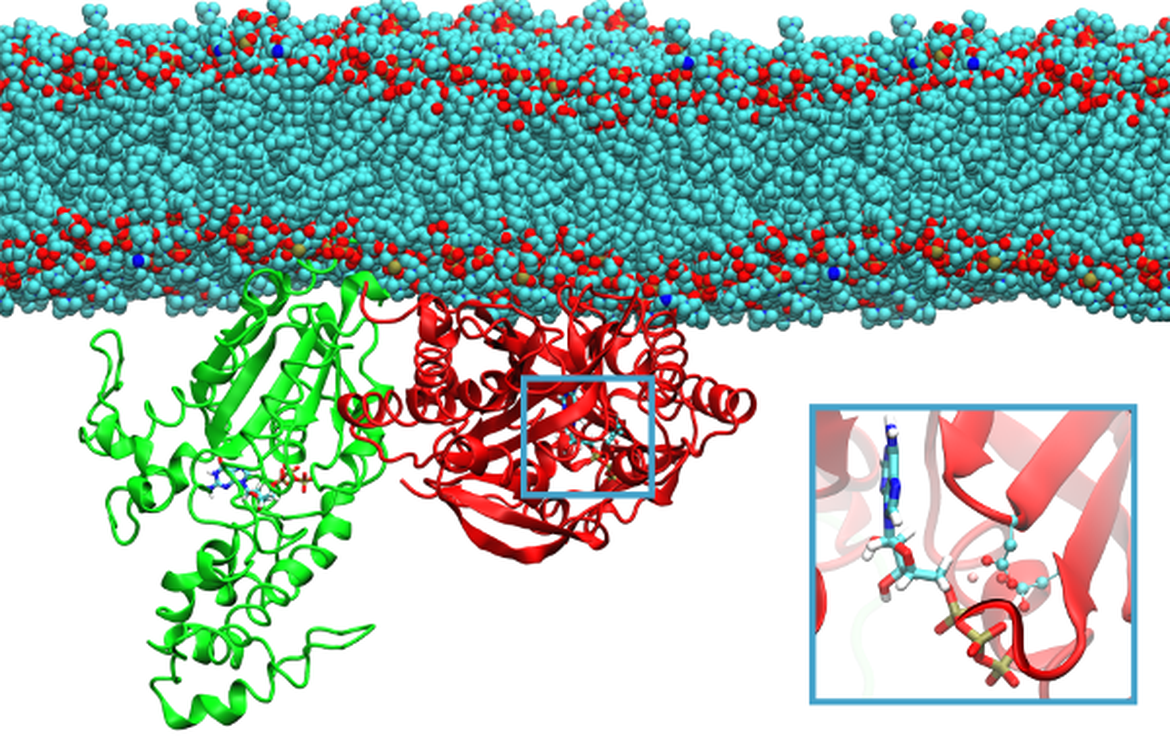
The response of these neurons is controlled by the plasma membrane-bound protein adenylyl cyclase type 5 (AC5), which controls the production of the secondary messenger cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP) that, in turn, stimulates protein kinase A (PKA), driving the downstream effects.
We are using molecular modelling methods to investigate the structure and activity of AC5, in the presence of regulatory proteins. As a proof-of-concept, the data we are generating is being used to constrain kinetic models of signalling cascades (see Molecular Signalling Cascades).
We are also exploring the binding processes of small organic compounds to neuroreceptors and building models for the prediction of the kinetic and thermodynamic properties of such complexes. Specifically, we consider the human M2 muscarinic acetylcholine receptor(mAchR), which is involved in psychiatric disorders and neurological diseases, such as Alzheimer's and Parkinson's diseases. In our study, we build a model that helps us to understand the ligand recognition mechanism and its connection with the protein conformational dynamics, the effects of allosteric modulators and other factors, such as membrane composition, that may affect binding thermodynamic and kinetics.
What is our specific take?
There are nine membrane-bound isoforms of mammalian adenylyl cyclase, whose catalytic unit is formed as a dimer of two domains. The 3D structure of the adenylyl cyclase catalytic unit, formed by domains from two different isoforms and in complex with its stimulatory G protein subunit, has been resolved by X-ray crystallography. However, its structure when bound to the inhibitory G protein subunit is unknown. Additionally, it is not known if both the stimulatory and inhibitory G protein are able to bind simultaneously, or if the resulting ternary complex would be active or inactive. It was previously shown that a lipid modification of the inhibitory G protein was required in order for it to modulate AC5 activity, however the effect of this modification on structure was not known.
To investigate this further, we have created models of all nine AC isoforms, and compared their electrostatic properties to known isoform-specific regulations patterns by interacting proteins (Tong et al., 2016). This allowed us to confirm the previously suggested binding position of the inhibitory G protein on AC5. The effect of lipid modification of the inhibitory G protein on its structure was investigated using modelling and simulation techniques (van Keulen and Roethlisberger, 2016), and the resulting structure docked to AC5 to study the mechanism of its effect on AC5 activity (van Keulen and Roethlisberger, 2017). These data, along with further simulations have been used to constrain a model of AC5-dependent signalling (see Molecular Signalling Cascades).
In the study of molecular recognition by neuroreceptors, we are applying a range of enhanced sampling molecular dynamics simulation approaches to explore ligand unbinding dynamics, egress routes and energy landscapes. We have investigated the dissociation of the complex of mAchR embedded in a neuronal membrane with the compound iperoxo, reconstructing the full free energy landscape of the ligand binding/unbinding process using funnel metadynamics simulations.
We have also employed the tauRAMD approach (Kokh et al., 2018), which enables efficient, extensive and unbiased sampling of possible egress paths of the compound from its binding site on the receptor, as well as the estimation of relative residence times. We have applied the method to two G-protein coupled receptors (GPCR), one of which is mAchR. We are simulating the unbinding of Iperoxo from mAchR and investigating the effect of allosteric ligand binding and different membrane compositions on this process.
Roadmap
In 2019, we published a constrained model of adenylyl cyclase signalling (Bruce et al., 2019, see Molecular Signalling Cascades).
We carried out studies to develop methods to model and simulate molecular binding and to apply these approaches to predict and understand molecular binding events. Protein binding sites can be identified with ArDock (Reille et al. 2018) and protein dynamics studies studied with internal coordinate normal mode analysis (Frezza et al. 2019) and the effects of protein dynamics on binding site druggability investigated with TRAPP (Yuan J-H et al., 2020).
In Spring 2019, we published a new model that enables reconstruction of a full-energy landscape of the agonist, iperoxo, binding to its target human neuroreceptor, the muscarinic acetylcholine receptor M2, embedded in a neuronal membrane. We have then evaluated in silico of the ligand’s residence times of iperoxo (Capelli et al, JCTC 2019). We have also investigated the limitations of current biomolecular force fields in calculating this quantity. The paper will be submitted shortly.
Finally, we have published a new methodology particularly useful to investigate different pathways of ligands in receptors (Capelli et al, JCP Lett 2019, Bonomi et al, Nature Methods 2019)
We have established protocols for the application of the tauRAMD method to membrane-bound receptor proteins to compute the residence times of their ligands. We are working on their application to studying ligand-GPCR dissociation under different conditions (presence or allosteric ligand, membrane composition, conformation).
We are aiming at developing user-friendly tools that will reveal the main determinants of ligand unbinding processes, providing guidelines for ligand optimization, and for understanding allosteric, agonist and antagonist effects. For this purpose, we have developed a machine learning analysis of simulated tauRAMD trajectories to decipher the molecular determinants of drug-target residence times (Kokh et al. 2019). Details and relevant data are provided in a live paper.
Many of the methods to compute binding kinetics are described in KBbox (Bruce, Ganotra et al.2019) which is an on-line toolbox designed to guide a new user in choosing an appropriate method for his or her problem.
Who's involved?
The Human Brain Project takes a multiscale approach to modelling the brain, and even when working at the smallest of spatial scales, that of individual molecules, a multiscale effort is needed to accurately model the molecular processes that occur within a neuron.
To allow us to study reactions of how enzymes are able to convert their substrate molecules into their products, we require quantum mechanical techniques that are able to model chemical reactions. Atom-resolution molecular dynamics simulations are used to investigate the internal motion of proteins, and how proteins interact with each other to modulate their activity. Coarse-grained molecular dynamics simulations can reach much longer timescales, and be used to model much larger multiprotein assemblies, allowing us to simulate processes that would not be feasible with atomically detailed simulations. Brownian dynamics simulations offer a way to model how proteins are able to diffuse within a neuron to find their interaction partners and can simulate timescales way beyond those accessible in the molecular dynamics simulations. We combine these simulation methods with a variety of bioinformatics and molecular modelling techniques.
The following people and their teams are driving this effort:
Paolo Carloni, Forschungszentrum Jülich, Germany
Richard Lavery, Centre National de la Recherche Scientifique, Lyon, France
Juliette Martin, Centre National de la Recherche Scientifique, Lyon, France
Modesto Orozco, Institute for Research in Biomedicine, Barcelona, Spain
Ursula Röthlisberger, École Polytechnique Fédérale de Lausanne, Switzerland
Rebecca Wade, Heidelberg Institute for Theoretical Studies, Germany
Nuria Cirauqui Diaz, Centre National de la Recherche Scientifique, Lyon, France
Francesco Colizzi, Institute for Research in Biomedicine, Barcelona, Spain
Elisa Frezza, Université Paris Descartes, France
Adam Hospital, Institute for Research in Biomedicine, Barcelona, Spain
Daria Kokh, Heidelberg Institute for Theoretical Studies, Germany
Guillaume Launay, Centre National de la Recherche Scientifique, Lyon, France
Aurelien Luciani, Institute for Research in Biomedicine, Barcelona, Spain
Daniele Narzi, École Polytechnique Fédérale de Lausanne, Switzerland
Benefit to the Community
- We are making raw simulation data available via the Brain Simulation Platform, allowing researchers to further analyse these in their own work.
- We are providing software and workflows that will allow the methods we have applied to be used by others in the neuroscience community. At present, the available tools include the Brownian dynamics simulation package SDA, its webserver webSDA, the protein-protein binding site prediction webserver ArDock and the library of molecular dynamics simulations MoDEL.
- We are currently constructing a new highly parallel quantum mechanical/classical mechanical software code, that can be used to describe enzymatic reactions. This is able to optimally use the large supercomputers which are becoming increasingly available to EU users. Indeed, the EU is greatly investing in powerful highly parallel computer architectures, and our developed code is particularly well suited for this.